Essential Content/Background
This is a multi-step curriculum. In this unit, I cover nearly a fourth of my overall curriculum in order for students to engage and explore over the full range of biological knowledge. Below is an outline for the curriculum.
Cell Biology
This portion of the curriculum will involve learning the basic components of DNA. Students will learn the history of the discovery of DNA and the methods used to produce images of its structure. Students will learn of the Central Dogma to see the role of DNA as it is replicated, transcribed and translated into proteins. Once students demonstrate proficiency in the expression and function of genes, students will learn about the types of mutations that cause disease by changing the characteristics of the DNA.
The human body is one organism, composed of a set of organs that provide essential functions, made up of layers upon layers of tissue, constructed from a specific grouping of cells. [1] We are a unique mosaic of eukaryotic cells that contain within them the intricate mechanisms for sustaining life. Organelles of varying structure serve a number of functions that control the integrity and life of the cell, which in turn keeps the body healthy and functioning properly. It's incredible to consider all the processes that occur within a microscopic cell. Nestling safe within all 50 trillion cells of the human body is our genetic blueprint, safe in the porous membrane of the nuclei that contain DNA.
Not too long ago, approximately 60 years ago, one of the greatest discoveries in science occurred, changing the way scientists view the entire field. This is not to be taken lightly. The exponential growth in the study of DNA since it's discovery is remarkable. Within the 20 th century, scientists not only discovered the blueprint for life (DNA) but also were also able to determine the structure and characteristics to its structure. This breakthrough in science made a huge affect on the research and answers that followed. James Watson and Francis Crick in 1954 made the breakthrough discovery of an era, defended by the X-ray diffraction images developed by Rosalind Franklin, who is often overlooked as an essential contributor to the discovery of DNA. [1] DNA stands for deoxyribonucleic acid, which carries the genetic information for each individual. From one person to the next, our genetic information is nearly identical, however it's in the small differences between our DNA that makes us appear different. Each of our 50 some trillion cells that make our bodies carries the entire genome of a human. Compacted within every nucleus of a cell are 23 pairs of chromosomes that contain genetic material. If the DNA of our chromosomes from a single cell were to be laid out in a line, it would reach nearly 20 meters in length.
Considering the importance of DNA, one might think that its structure is quite elaborate, however if you can picture a ladder, the structure is quite simple to grasp. DNA has a double helical structure, also referred to as a twisted ladder model. Two anti-parallel strands with a sugar-phosphate backbone outline the long stretch of genetic material. [2] The rails, or steps, of this ladder are constructed of nitrogenous bases (A, C, T, G), two bases per rail, with the paired bases held together with hydrogen bonds.
Figure 1
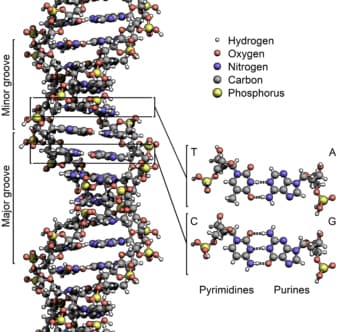
wikipedia.org
Genetics
DNA is constantly replicating through cell division. Within the small confinement of the nucleus, replication occurs producing additional strands of DNA to insure the longevity of the organism. From duplicated DNA strands, a process called the Central Dogma beings. Refer to figure 2. This process takes DNA that has been replicated and transcribes the strand into mRNA using the nitrogenous bases (A, C, U, G). As you may notice, the thymine (T) of DNA is now substituted for uracil (U) in RNA. The strand of RNA leaves the nucleus and enters the cytoplasm of the cell until it locates a free ribosome at attaches to it. At the ribosome, codons (discussed below) pair with anti-codons that transport an amino acid. A combination of amino acids, and a specified folding of the strand, determines the translation of a protein that has specific functions in the organism.
Figure 2
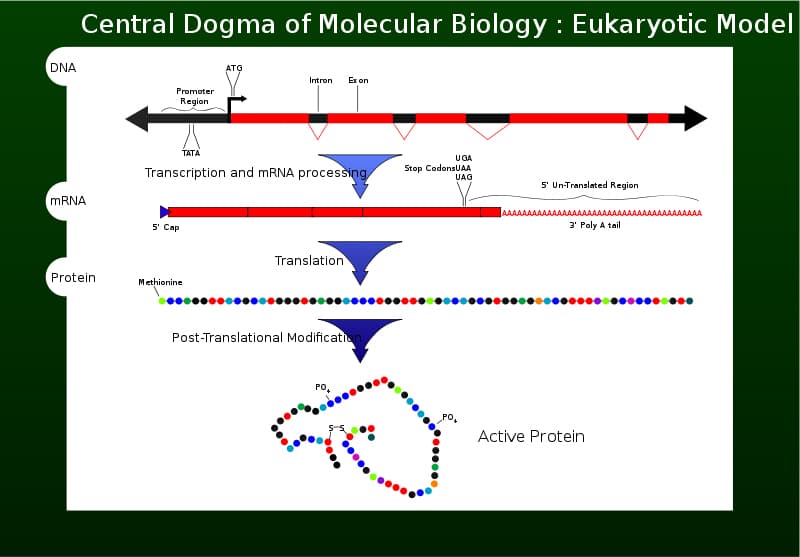
wikipedia.org
Codons are a set of three adjacent bases on mRNA that code for an amino acid. Since there are 4 bases in each position, there are 64 possible codons available to code for 20 different amino acids. [2] Varying amino acids come together to make proteins and each protein has its own specific function. The expression of proteins is responsible for our traits, both physical and at the cellular level. Certain proteins have vital responsibilities in the upkeep of an organism, while others can be harmful. We will be examining the harmful affects of proteins in the health section of this unit. The genetic code is a table that is used to determine the codons that represent a specific amino acid. In Figure 3 below, you can see that the position of the first, second, and third bases correlate with an amino acid.
Figure 3

wikipedia.org
In the DNA a set of nitrogenous bases code for a gene. This gene goes through the Central Dogma process to find its way to producing its protein. It is a miraculous organism, the human body, but it is not without faults. You can relate to that. For instance, we add numbers on a daily basis. It is not a complicated task; it almost feels mind numbing. However, we sometimes make mistakes. Even the simplest mistake can produce an absolute wrong answer. Our DNA functions very much like we do. Although it DNA is replicated continuously through life, as we replace some of our trillions of cells, it is prone to mistakes, referred to as mutations. There are a number of possible mutations including substitution, insertion, deletion and frameshift. [3]
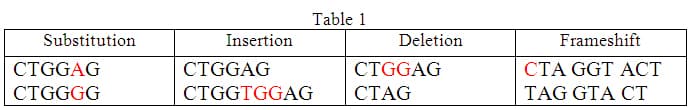
Substitutions are simple to identify in table 1 above. These mutations are easy to comprehend but not so easy to identify in DNA. A single nitrogenous base is substituted for another. If the base that is substituted codes for the same amino acid as the original, the mutation is considered silent. Although this change is subtle, it can lead to the expression of mutated proteins, such as in sickle cell anemia, discussed in detail below. Some times when mutations occur, it can change the amino acid that is next in line, for instance if the substitution prematurely signals a STOP codon, an incomplete protein will be produced: the incomplete protein likely won't function as it should. Insertions are additional bits of DNA added to the existing strand. In the above example, TGG is added making the initial strand longer. Deletion is caused by the loss of bases from the strand. Lastly, frameshift may seem similar in concept to deletion however the differing factor is that the deletion that occurs shifts every proceeding frame of DNA causing the translation of an entirely different protein that originally instructed. Other types of mutations exist, but for the purpose of this unit I will only address these four.
Physiology (Disease and Human Health)
The design challenge for students will be based on 4 different diseases: Huntington's Disease, Sickle Cell Anemia, Hemophilia and Cystic Fibrosis. These are chosen because each are caused by a single gene mutation and are relevant to many in my student population. My students know Sickle Cell Anemia and Hemophilia because many of their family and friends either have or know someone who has it. The students are rather interested in these two in particular and it would be great for them to learn more while completing the assignment.
The first of the single gene diseases that will be studied is Sickle Cell Anemia. It is a condition that produces deformed red blood cells (RBCs), which are essential for oxygen transportation throughout the body. The disfigured sickle (crescent) shape of the cell keeps the cells from adequately transporting oxygen to organs and tissues. Red blood cells contain hemoglobin, which is a protein that is responsible for the transport of oxygen. Abnormal hemoglobin is found in sickle cells, which determines the sickle shape of the cell. Unlike the circular donut shape of normal RBCs, this sickle shape causes the cell to be rigid and sticky. This keeps blood from moving freely throughout the vessels and causes backed up and blocked blood flow, often resulting is severe pain and organ damage for the individual. Sickle cells also have a much shorter life span of 10-20 days compared to that of normal RBCs, which live for nearly 120 days in the circulation. [4] Because of this short life span, it is difficult for the bone marrow to produce new RBCs fast enough to replace the ones that are disappearing: this causes a decreased RBC count, or anemia. Sickle Cell Anemia is an inherited condition that requires both parents to pass the trait to their offspring in order for it to be expressed.
People with sickle cells experience chronic pain and fatigue. Unfortunately, there is no cure for sickle cell but sufferers can be closely monitored and cared for to control their symptoms. Sickle Cell is a common disease among those with ancestors from Africa and it affects millions of people worldwide. In America, sickle cell affects somewhere between 70,000 to 80,000 individuals and roughly one in every 500 African Americans and one in every 1,000 Hispanic Americans. [4] Sickle Cell Anemia occurs from a mutated gene known as HBB located on chromosome 11. Healthy hemoglobin has four units, two known as alpha-globin and two known as beta-globin. Mutations of the HBB gene prevent growth of adequate beta-globin numbers. The HBB gene results in a number of different mutations, in sickle cells: in particular, both beta-globin units are replaced with HbS (hemoglobin S). [4] This mutation leads to the short life of sickle cells and inefficient transport of oxygen discussed above.
Huntington's Disease is the next single gene disease. It is a brain disorder that causes uncontrolled movements, emotional instability and degeneration of cognition. The inherited gene usually remains silent through adolescent years and begins to show signs when a person reaches their thirties or forties, which is referred to as adult-onset. In the beginning symptoms are mild and less frequent, they include irritability, depression, small involuntary movements, lack of coordination and cognitive troubles. [5] Huntington's disease is most recognized by jerking movements or twitches known as chorea, which is where Huntington's gets its other name of Huntington's Chorea. When the disease has progressed, inflicted individuals experience great difficulty in mundane physical tasks such as walking and their speech becomes slurred and incomprehensible. From the first clinical signs of Huntington's affected individuals have a life expectancy of about 15-20 years. Another less common form of Huntington's disease is referred to as juvenile and begins in childhood and early adolescence. The signs and symptoms are similar to that of adult-onset Huntington's but, in this case, 30-50% of individuals affected experience seizures as a result of the disease. Unfortunately, with juvenile Huntington's individuals live about 10-15 years from onset. In contrast to Sickle Cell Anemia, Huntington's disease is most common among those of European ancestry affecting approximately seven people in 100,000. [5]
Huntington's disease is an inherited condition that only requires inheritance from one parent for it to be expressed because it is an autosomal dominant trait. In other words, the trait for Huntington's disease is dominant; therefore just one allele with the trait and the offspring will be affected. HTT is the gene found on chromosome 4 that correlates with Huntington's disease. The normal function of the HTT gene has not been found but it is known to contribute to the function of the nervous system. From the HTT gene a protein called Huntingtin is formed. The mutation has very specific characteristics involving only the bases adenine, cytosine, and guanine in a trinucleotide repeat. In a normal gene the bases CAG are repeated a short 10-35 times; however, in people with Huntington's disease CAG repeats 36-120 times in a row resulting in a larger strand of DNA. [5] Those with juvenile Huntington's tend to have longer strands (<60) of the repeating nucleotides whereas adult onset tends to be between 40 and 50 repeats. The shorter an individuals repeats of bases CAG of their HTT gene the less likely they will have Huntington's and the less possibly of passing it on to their offspring. The elongated strand of CAG repeats causes a mutated huntingtin protein which may have a role in disrupting normal neural functions that characterized Huntington's disease.
Hemophilia is a bleeding disorder in which the clotting time of blood is slowed, causing those affected to bleed excessively and for prolonged periods of time. Simple cuts can require immediate medical attention because of the risk of too much blood loss. For more severe forms of hemophilia there is even risk of spontaneous unprovoked bleeding that leads to more serious complications. There two types of hemophilia: Hemophilia A, the most common type, and hemophilia B. Both exhibit the similar signs and symptoms but they are caused by mutations in different genes. Controlled blood clotting is necessary to keep the body from losing too much blood. Without the body's natural ability to control blood clotting, it is crucial to be aware of the condition. For the purpose of this unit, focus will be on hemophilia A. The gene corresponding with hemophilia is found on the X chromosome (sex chromosome 23). Hemophilia A occurs in 1 in 4,000 males worldwide, with hemophilia B occurring in 1 in 20,000 newborn males. [6]
As mentioned above, the gene for hemophilia is located on chromosome 23. It is an inherited X-linked recessive disorder. Since the trait is only carried on the X chromosome, inheritance of one altered copy of the gene is sufficient enough to cause the condition. If a female were to get one altered and one wild type of the gene, she will be unaffected, therefore hemophilia is less common among females. On the X chromosome the gene F8 is located and has the blueprint for synthesizing a protein called coagulation factor VIII. [6] Coagulation factors are essential in the blood clotting mechanism. Mutations of the F8 gene results in production of the coagulation factor VIII that reduces the amount of proteins available which is the cause of the disorder. A lower coagulation factor count correlates with a more severe condition.
Cystic Fibrosis is the last of the single gene diseases this unit covers. It is an inherited life-threatening condition affecting a number of systems in the human body. It is the most common lung disease in children and young adults. Thick, sticky mucous builds up in the lungs and digestive tract due to an abnormal transport of chloride and sodium across tissue. Severe difficulty breathing and dangerous lung infections are the most common conditions due to this disorder. These symptoms can be treated with antibiotics and other medications, but treatment is rarely complete. Cystic fibrosis is most common among Caucasians, affecting roughly one in every 3,000 individuals. [7] Fetuses and infants are tested for CF so proper precautions and necessary transplants can be carried out.
Cystic fibrosis is another autosomal recessive gene, meaning a child would have to receive a recessive allele from both parents in order for the condition to be expressed. The gene for cystic fibrosis is found on chromosome 7: its official name is transmembrane conductance regulator (CFTR). [7] The protein made from this gene, which creates chloride channels, is responsible for the maintenance of normal mucous secretions that reside on epithelial tissue. Viscosity of secretions is regulated by the normal chloride transport, keeping it thin and permeable. Any change in the CFTR gene will produce proteins that are insufficient to carry out their chloride transport function. There are thousands of possible mutations that leads to the production of CF. They differ in the system it affects. Some individuals experience more complications with the respiratory system and other with the gastrointestinal system. Since CF affects secretions of the cells throughout the body, complications generally occur throughout. Some mutations of CFTR are very mild, allowing CFTR to maintain some its function. But severe forms involve only small changes in CFTR. For example, a single amino acid change or deletion in the CFTR gene can result in channel proteins that do not reach the cell membrane and therefore cannot properly transport chloride ions.
Biotechnology
Students will explore different methods to read DNA to identify specific genes and compare the genes of specific offspring with their parents. Specifically, students will learn about Gel electrophoresis and how it is used to test for the Huntingtin Gene. Students will conduct a lab using gel electrophoresis apparatuses and fake blood samples.
Gel electrophoresis is a technique used to determine lengths of particular strands of DNA. Gel electrophoresis does not provide detailed information about the particulars of a DNA strand however the length of a strand can provide relative information that is useful in genetic testing. DNA has a negative charge due to the phosphate group in the rails of the double helix. Gel electrophoresis exploits the negative charge of DNA: an electrical field in an apparatus drives the DNA towards the positive end. [1] DNA travels through an agarose gel bed that can be made with different concentrations to change the density of the matrix within the gel. The higher concentration of matrix, or webbing, in the gel, the more confined larger strands of DNA are, thus hindering them from moving. Short strands of DNA can move more freely down the gel. Gel electrophoresis is useful when determining the presence of large strands of DNA caused by large genes, such as HTT, which is a marker for Huntington's disease. Information from gel electrophoresis is amplified with the use of PCR (Polymerase chain reaction) a process that produces thousands of copies of the particular DNA strand. [7] Many copies allows researchers to compare the DNA across several magnitudes to collect more solid information about the gene present.
Cells in our body each contain all or our genes, but only a small portion of those genes will be expressed in a cell. All organs and tissues have specific functions and the cells in that tissue will express only the genes necessary for proper function. Synthesizing proteins from DNA requires transcription factors that bind to specific regions that initiate the transcription of the gene. [8] If a gene is not transcribed than the protein is not made and the gene is not expressed. Gene control can also occur during RNA transcription by creating different splices of mRNA. The differences in mRNA will alter the expected protein. These differences can cause inactivation of the protein desired. Blocking the expression of a gene can potentially have the ability to silence the expression of a mutated gene; at least this is an idea that students will ponder on.
Blocking of gene expression can occur in a number of ways. There are single stranded antisense oligonucleotides with a short life span that can be used to bind to base pairs in DNA to block transcription or RNA to block translation. There are also small interfering RNAs (siRNA) that are used to target degradation of specific mRNA transcripts. [8] This process allows for the gene in the DNA to be expressed (i.e. transcribed to mRNA) however at the transcription phase siRNA can silence mRNA and therefore turn off protein production. This method could potentially be helpful in treatment of Huntington's disease. These methods of gene therapy are relatively simple to comprehend but it is not without its limitations. This offers great room for students to consider possibilities and create methods that seem plausible to cure their studied disease.
This section of the content is tied in with moral and ethical discussions related to genetic testing, screening and engineering. Are these processes useful to mankind? Harmful? What implications can be made considering the growth and use of genetic screening in determining life? What does it mean for human evolution?
Comments: